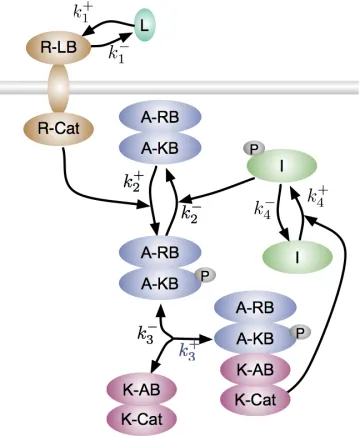
Our central research goal is to understand the evolution and function of the complex molecular networks that underlie life. Our research program thus integrates population genomics and systems biology toward a long-term goal of incorporating molecular mechanisms into evolutionary genetics.
Population genomics
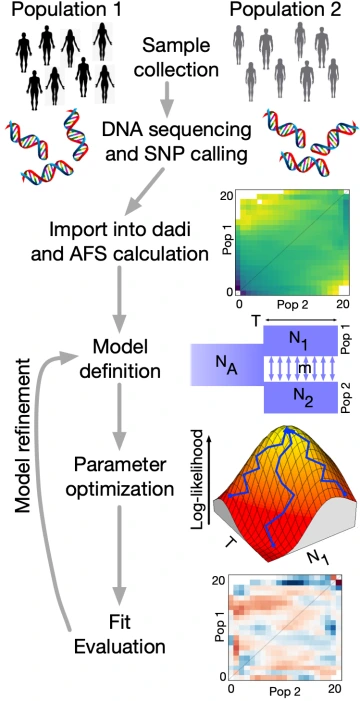
We have unique expertise in numerical diffusion approaches for modeling population genomic data, stemming from Ryan’s development of the software dadi (Diffusion Approximations for Demographic Inference). For example, we developed a diffusion approach for inferring divergent selection on mutations among populations (Huang et al. Mol Biol Evol 2021).
We strive to make population genomics inference approaches broadly useful to the research community. For example, we showed that the Godambe Information Matrix provides an effective and computationally efficient approach to statistical analysis of population genetics models (Coffman et al. Mol Biol Evol 2016). We are currently running the Genomic History Inference Strategies Tournament (Struck et al. Mol Biol Evol 2025), an effort to evaluate inferences methods through community competition. We are also active contributors to the stdpopsim project aiming to provide standardized simulation resources to the community (Lauterbur et al. eLife 2023).
We are also exploring modern machine learning methods for population genomics inference. We recently developed an approach for demographic history inference based on neural networks trained on site frequency spectra (Tran et al. Mol Biol Evol 2024). We also developed a method for evaluating the general performance of machine learning methods trained on haplotype matrices (Tran et al. Mol Biol Evol 2025).
Cancer
Cancer is fundamentally an evolutionary process, as tumor cells acquire mutations to enhance their growth, outcompete their neighbors, and evade the immune system. We thus apply concepts and tools from population genomics to understand tumor development.
Tumor genomic sequencing data are critical for understanding genetic variation within tumors, which affects their development and recurrence. In particular, low frequency mutations within tumors are strongly informative about tumor development, but they are difficult to identify among sequencing errors. To improve identification, we developed a Bayesian approach that incorporates the fact that every tumor has a unique spectrum of mutational types that it tends to generate (Mannakee and Gutenkunst NAR: Genomics and Bioinformatics 2020). High-frequency variants can be used to identify this spectrum, so it can be applied to improve identification of low-frequency variants, enabling more accurate evolutionary inferences.
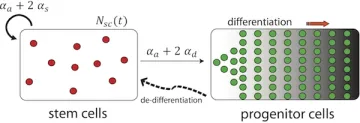
A key goal of cancer evolution research, with applications to cancer prevention, is to understand the factors that hasten the acquisition of tumor driver mutations. Tumors can quickly acquire driver mutations by elevating their mutation rate, but not all tumors do this. In theoretical work, we established an alternative possibility, that tumors may benefit from altering not the total rate of mutations but rather the spectrum of types of mutations (Tuffaha et al. Am Naturalist 2023). Using data from thousands of tumors, we empirically validated this prediction (Tuffaha et al. Mol Biol Evol 2025). Our analysis also suggested that hypoxia drives particularly potent changes in mutation spectra, a prediction we are following up on.
Mutations within tumors can not only promote growth, they can also generate novel antigens that provoke an immune response. By contrasting genomic data from tumors in immunocompetent versus immunocompromised patients, we recently identified key factors that identify immunogenic mutations (Borden et al. submitted). We are leveraging these insights to better identify tumor vaccine targets, and we are developing models to better understand the immune system’s effect on early tumor development.
Evolutionary systems biology
Our group also has unique expertise in integrating dynamical systems biology models of molecular networks with genomic data. A particular focus has been predicting rates of protein evolution. Knock-out experiments explain very little of the variation in evolutionary rates. This has led some researchers to doubt whether function plays much of a role in constraining protein evolution. We think knock-out experiments may be misleading, because knock outs are massive perturbations, and many evolutionarily-relevant mutations introduce only small perturbations. We use dynamical systems biology models to assess the effects of differentially small perturbations to molecular functions. Using this approach, we showed that proteins with greater influence on network dynamics evolve more slowly in a large number of networks (Mannakee and Gutenkunst PLoS Genetics 2016) and in yeast pyrimidine biosynthesis (Hermansen et al. BMC Evol Biol 2015).
Our modeling work is complemented by bioinformatic approaches. For example, we recently analyzed all human and yeast proteins in terms of solvent accessibility, structural disorder, expression level, and mutation frequencies to test the strength of selection against spurious phosphorylation by tyrosine kinases (Pandya et al. Mol Biol Evol 2015).